Optimizing workflows for freshwater metabarcoding
Written on June 10th, 2020 by Bilgenur Baloglu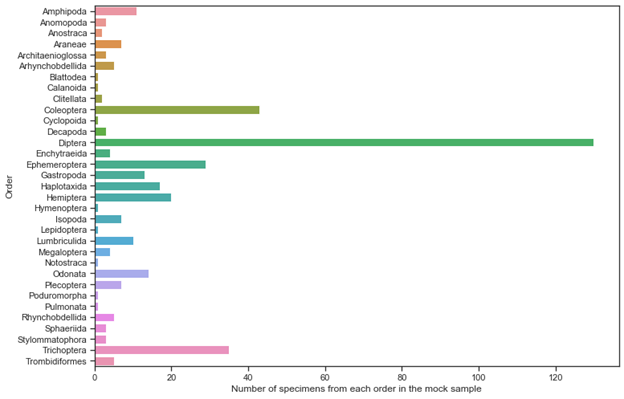
How can we improve metabarcoding workflow? By validating primers and skipping size sorting
Global freshwater biodiversity is declining more rapidly than the diversity of many stressed terrestrial ecosystems, e.g., 1–8% species loss per decade. Such loss of freshwater biodiversity affects food webs, nutrient cycling, climate, air quality, and water supply.The lack of efficient and rapid assessment tools has been a major drawback with monitoring the health of freshwater ecosystems, where most environmental samples oftentimes contain small-sized organisms that are lacking diagnostic characters or morphologically degraded.
Metabarcoding is a faster, cheaper and easier-to-use alternative to conventional environmental biomonitoring. The accuracy of metabarcoding studies is dependent on the recovery of the targeted taxa using amplification primers as well as sample preparation procedures. In this study, I have tested twenty-one published, nine newly combined, and one newly designed primer sets in silico and in vitro across a diverse set of 401 aquatic invertebrate species, representing 31 orders, to establish optimal protocols for Illumina Illumina MiSeq based metabarcoding of kick-net samples.
Bulk sample metabarcoding is useful if it can capture the entire diversity in a given sample. As organisms in a bulk sample may have vastly different biomasses, detecting rare and small species has been a challenge in metabarcoding studies based on the analysis of pooled DNA extractions. In this study, I have tested the effect of sample sorting by biomass using the primer sets on two mock samples, one sorted and one unsorted by specimen biomass.
Manuscript in progress.


